The Journal of Chemical Physics, 08 January 2005
J. Chem. Phys. 122, 024901 (2005) (18 pages)
©2005 American Institute of Physics. All rights reserved.
Previous section: TITLE PAGE
Next section: II. METHODS
I. INTRODUCTION
For the past ten years, there have been many attempts1–35 to develop coarse-grained scoring potentials that can identify native structures from non-native folds.36,37,38,39 These simplified potentials are useful in studies of protein structural prediction40,41,42,43 and protein dynamics and folding mechanism28,29,44 because it is computationally difficult to use all-atom molecular dynamics simulations for these purposes.
The idea of using residue-residue contact frequencies to represent contact preferences between amino acids was proposed first by Tanaka and Scheraga,1 and a contact potential2,3,4 for each type of amino acid pair at a residue level was evaluated in the Bethe approximation under the assumption that protein structures can be regarded as a mixture of disconnected residues in statistical equilibrium. Sippl8 introduced a distance dependency into a pair potential and evaluated it as a potential of mean force. Score functions at an atomic level were also devised.11,12,13,14,18 The capabilities of pairwise score functions to identify native structures from non-native folds have been examined by those optimizations,19,20,21,22,23,24,25 and it was reported that it is impossible to make a pairwise potential21 and even a distance-dependent potential23,24 to identify all native structures. Multibody potentials have also been derived and the importance of multibody interactions have been pointed out.28,29,30,31 Liwo et al.32 developed a general method to derive multibody terms in a potential of mean force.
On the other hand, the importance of specific coordinations between residues in protein structures was pointed out by Bahar and Jernigan.45 Liwo et al.15,16 developed a united-residue force field that is both radial and anisotropic. The united-residue force field was determined by parameterizing physically reasonable functional forms of potentials of mean force for side chain interactions. Each side chain was represented by an ellipsoid and the relative orientation between side chains was described by three angles. The interactions between side chains were parameterized as van der Waals potentials. Buchete et al.34,35 also attempted to develop anisotropic statistical potentials from the observed distribution of relative residue-residue orientations in known protein structures. To represent the orientation of one residue relative to another, three degrees of translational freedom and three degrees of rotational freedom must be specified. A polar coordinate system and Euler angles can be used to specify the three degrees of translational freedom and the three degrees of rotational freedom, respectively. In their potentials, only radial distance and polar angle dependencies of relative residue-residue orientations are taken into account but Euler angle dependences of the orientations were not explicitly taken into account, probably because of the limited size of samples. Onizuka et al.33 attempted to estimate a fully anisotropic distance-dependent potential, which is a function of radial distance, polar, and also Euler angles, for each type of residue pair, although they could not achieve any improvement in the discrimination power of their score function by taking account of Euler angle dependencies. These analyses indicate the importance of residue-residue orientations in residue-residue interactions.
Here the fully anisotropic distributions of relative orientations between contacting residues are estimated as a function of polar and Euler angles from known protein structures. Those Euler angle dependencies and correlations between polar and Euler angles are analyzed as well as polar angle dependencies.
For evaluation of the frequency distribution of residue-residue orientations, we did not use a method of dividing space into many cells and counting samples observed in each cell, but instead employed the method proposed by Onizuka et al.33 in which the observed distribution of residue-residue orientations is represented as a sum of 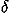 functions each of which represents the observed location in angular space, and then is estimated in the form of a series expansion with spherical harmonics functions, ignoring high frequency modes that occur, because of the sample size. High frequency modes are statistically less reliable than low frequency modes. Here, unlike other works33,34,35 each expansion term is separately corrected for the sample size according to suggestions from an analysis of Bayesian statistics. As a result, many expansion terms can be utilized to evaluate orientational distributions. A local coordinate system for each residue is defined for fold recognition, based only on main chain atoms to represent directional and rotational relationships between the main chains of contacting residues rather than between the side chains.33,34,35 Results show that a large contribution to the orientational entropy of residue pairs comes from the Euler angle dependencies of the frequency distribution and also from the polar and Euler angle correlations. Then, an energy potential for relative orientations of contacting residues is evaluated for each type of amino acid pair as a potential of mean force from the estimated distributions.
functions each of which represents the observed location in angular space, and then is estimated in the form of a series expansion with spherical harmonics functions, ignoring high frequency modes that occur, because of the sample size. High frequency modes are statistically less reliable than low frequency modes. Here, unlike other works33,34,35 each expansion term is separately corrected for the sample size according to suggestions from an analysis of Bayesian statistics. As a result, many expansion terms can be utilized to evaluate orientational distributions. A local coordinate system for each residue is defined for fold recognition, based only on main chain atoms to represent directional and rotational relationships between the main chains of contacting residues rather than between the side chains.33,34,35 Results show that a large contribution to the orientational entropy of residue pairs comes from the Euler angle dependencies of the frequency distribution and also from the polar and Euler angle correlations. Then, an energy potential for relative orientations of contacting residues is evaluated for each type of amino acid pair as a potential of mean force from the estimated distributions.
A reference state is also defined differently from other works.33,34,35 A reference distribution for each type of amino acid pair is the uniform distribution rather than the overall distribution for all types of amino acid pairs employed by other works,33,34,35 so that residue-residue orientations can be fully evaluated. The overall distribution may be one of the important characteristics to distinguish proteinlike structures from others, because the overall distribution observed in native structures is not known to be characteristic of non-native conformations. The zero energy level of the orientational potential for each residue pair type is defined such that the expected value of orientational energy for the native folds is equal to zero for each type of contacting residue pair. Therefore, this orientational potential represents simply the suitability of a given relative orientation between contacting residues. Also, this orientational potential can be used without any modification as a scoring function for optimum sequence designs and sequence-structure alignments in which deletions and additions of amino acids are allowed.7
It is shown that the discrimination performance of the orientational potential in fold recognition is significantly improved by taking account of Euler angle dependencies and the performance of a total energy potential consisting of a long-range contact potential and a short-range secondary structure potential is improved by taking account of the orientational potential as an additional term.
Previous section: TITLE PAGE
Next section: II. METHODS