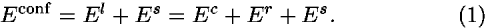
The short-range potential is a secondary structure potential based on peptide dihedral angles. All of these potentials are estimated as potentials of mean force from the observed distributions of residue-residue contacts and of peptide dihedral angles at the residue level in crystal structures of proteins. In the following, energy is represented in kBT units, where kB is the Boltzmann constant and T is temperature.
![<i>E</i><sup><i>c</i></sup> = (1/2) [summation]<sub><i>i</i></sub>[summation]<sub><i>j</i> [not-equal] <i>i</i></sub><i>e</i><sup><i>c</i></sup>(<b>r</b><sub><i>i</i></sub>,<b>r</b><sub><i>j</i></sub>),](024901_1-div2_files/024901_1m1.gif)
where ec(ri,rj) is the contact energy between the ith and jth residues, and ri represents all the atomic positions of the ith residue. The pairwise energy potential is represented as the sum of two terms, one of which is the usual contact potential2,3,4 and the other is a potential of mean force for relative orientations between contacting residues that is evaluated here from the statistical distribution of relative orientations,
![<i>e</i><sup><i>c</i></sup>(<b>r</b><sub><i>i</i></sub>,<b>r</b><sub><i>j</i></sub>) = <i>Delta</i><sup><i>c</i></sup>(<b>r</b><sub><i>i</i></sub>,<b>r</b><sub><i>j</i></sub>)[<i>e</i><sub><i>a</i><sub><i>i</i></sub><i>a</i><sub><i>j</i></sub></sub><sup><i>c</i></sup> + <i>e</i><sub><i>a</i><sub><i>i</i></sub><i>a</i><sub><i>j</i></sub></sub><sup><i>o</i></sup>(<b>r</b><sub><i>i</i></sub>,<b>r</b><sub><i>j</i></sub>)],](024901_1-div2_files/024901_1m2.gif)
where 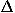 c(ri,rj) represents the degree of contact between the ith and jth residues, e
c(ri,rj) represents the degree of contact between the ith and jth residues, e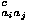 is the contact energy for residues of types ai and aj in contact, and eoaiaj(ri,rj) is the orientational energy for the relative direction and rotation between amino acids of type ai and aj contact; ai means the amino acid type of the ith residue. Here, it should be noted that the radial distance between residues is described by specifying whether or not these residues are in contact with each other, and that orientational interactions are assumed only for residues that are in contact with each other.
is the contact energy for residues of types ai and aj in contact, and eoaiaj(ri,rj) is the orientational energy for the relative direction and rotation between amino acids of type ai and aj contact; ai means the amino acid type of the ith residue. Here, it should be noted that the radial distance between residues is described by specifying whether or not these residues are in contact with each other, and that orientational interactions are assumed only for residues that are in contact with each other.
c(ri,rj) takes a value one for residues that are completely in contact, the value zero for residues that are too far from each other, and values between one and zero for residues whose distance is intermediate between those two extremes, about 6.5 Ĺ between geometric centers of their side chain heavy atoms. Previously, this function was defined as a step function for simplicity. Here, it is defined as a switching function as follows; in the equation below to define residue contacts, ri means the position vector of a geometric center of side chain heavy atoms or the C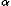 atom for GLY,
atom for GLY,
![<i>Delta</i><sup><i>c</i></sup>(<b>r</b><sub><i>i</i></sub>,<b>r</b><sub><i>j</i></sub>) [equivalent] 1.0,–<i>S</i><sub><i>w</i></sub>[|<b>r</b><sub><i>i</i></sub> – <b>r</b><sub><i>j</i></sub>|,<i>d</i><sub>1</sub><sup><i>c</i></sup>(<i>r</i><sub><i>a</i><sub><i>i</i></sub></sub><sup>vdw</sup> + <i>r</i><sub><i>a</i><sub><i>j</i></sub></sub><sup>vdw</sup>),<i>d</i><sub>2</sub><sup><i>c</i></sup>(<i>r</i><sub><i>a</i><sub><i>i</i></sub></sub><sup>vdw</sup> + <i>r</i><sub><i>a</i><sub><i>j</i></sub></sub><sup>vdw</sup>)],](024901_1-div2_files/024901_1m4.gif)
![<i>d</i><sub>1</sub><sup><i>c</i></sup>(<i>x</i>) [equivalent] {(6.5 × 0.95, for <i>x</i> <= 6.5 × 0.95; <i>x</i>, otherwise),](024901_1-div2_files/024901_1m5.gif)
![<i>d</i><sub>2</sub><sup><i>c</i></sup>(<i>x</i>) [equivalent] 1.05<i>d</i><sub>1</sub><sup><i>c</i></sup>(<i>x</i>)/0.95,](024901_1-div2_files/024901_1m6.gif)
![<i>S</i><sub><i>w</i></sub>(<i>x</i>,<i>a</i>,<i>b</i>) [equivalent] {(1 for <i>x</i> <= <i>a</i>; [(<i>b</i><sup>2</sup> – <i>x</i><sup>2</sup>)<sup>2</sup>/(<i>b</i><sup>2</sup> – <i>a</i><sup>2</sup>)<sup>3</sup>][3(<i>b</i><sup>2</sup> – <i>a</i><sup>2</sup>); – 2(<i>b</i><sup>2</sup> – <i>x</i><sup>2</sup>)] for <i>a</i> < <i>x</i> < <i>b</i>; 0 for <i>b</i> <= <i>x</i>; ),](024901_1-div2_files/024901_1m7.gif)
![<i>r</i><sub><i>a</i></sub><sup>vdw</sup> = [((3 <i>rho</i> <i>V</i><sub><i>a</i></sub>)/(4 <i>pi</i>))]<sup>1/3</sup>,](024901_1-div2_files/024901_1m8.gif)
![<i>rho</i> = ((<i>pi</i>)/(3[square root of 2])),](024901_1-div2_files/024901_1m9.gif)
where Sw is a switching function, and r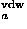 is the van der Waals radius of a residue of type a which is estimated from the average volume Va occupied by a residue of type a in protein structures with the packing density of hard sphere
is the van der Waals radius of a residue of type a which is estimated from the average volume Va occupied by a residue of type a in protein structures with the packing density of hard sphere 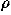 ; Va are those calculated in Refs. 46 and 47 and listed in Ref. 2. A critical distance to define a residue-residue contact is about 6.5 Ĺ, but it is taken to be larger for bulky residues.
; Va are those calculated in Refs. 46 and 47 and listed in Ref. 2. A critical distance to define a residue-residue contact is about 6.5 Ĺ, but it is taken to be larger for bulky residues.
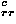 and a residue-type dependent term e
and a residue-type dependent term e![<sub><i>a</i><i>a</i><sup>[prime]</sup></sub><sup><i>c</i></sup>](024901_1-div2_files/024901_1m12.gif) ; r means an average residue here.
; r means an average residue here.
![<i>e</i><sub><i>a</i><i>a</i><sup>[prime]</sup></sub><sup><i>c</i></sup> = <i>Delta</i> <i>e</i><sub><i>a</i><i>a</i><sup>[prime]</sup></sub><sup><i>c</i></sup> + <i>e</i><sub><i>r</i><i>r</i></sub><sup><i>c</i></sup>.](024901_1-div2_files/024901_1m13.gif)
The energies e for all pairs of the 20 types of residues were recalculated44 from 2129 protein species representatives of the SCOP48 Release 1.53 with the sampling method3 and with the parameters evaluated in Miyazawa and Jernigan4 to correct these values estimated by the Bethe approximation; actually, the estimates of contact energies corrected for the Bethe approximation are divided by 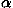
![[prime]](024901_1-div2_files/prime-script.gif)
 0.263 defined in Eq. (34) of that paper4 and used as the values of e. In other words, the intrinsic pairwise interaction energies
0.263 defined in Eq. (34) of that paper4 and used as the values of e. In other words, the intrinsic pairwise interaction energies 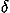 eij are corrected relative to the hydrophobic energies eir, and the hydrophobic energies are not corrected at all; see that paper4 for the exact definitions of eij and eir. This scheme is employed, so that all the energy potentials in Eq. (1) have magnitudes estimated as the potential of mean force from observed distributions by assuming a Boltzmann distribution.
eij are corrected relative to the hydrophobic energies eir, and the hydrophobic energies are not corrected at all; see that paper4 for the exact definitions of eij and eir. This scheme is employed, so that all the energy potentials in Eq. (1) have magnitudes estimated as the potential of mean force from observed distributions by assuming a Boltzmann distribution.
is essential for a protein to fold by canceling out the large conformational entropy of extended conformations but it is difficult to estimate.2,3 The value –2.55 originally estimated2,3 for e is used here; as a result, the contact energy e takes a negative value for all amino acid pairs except for LYS-LYS pair.
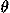 ,
,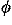 ) and Euler angles (
) and Euler angles (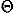 ,
,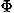 ,
,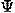 ) to describe the direction and rotation of one residue relative to another, respectively. A local coordinate system fixed on each residue will be defined later. The potential of mean force for residue orientations is defined as
) to describe the direction and rotation of one residue relative to another, respectively. A local coordinate system fixed on each residue will be defined later. The potential of mean force for residue orientations is defined as
![<i>e</i><sub><i>a</i><i>a</i><sup>[prime]</sup></sub><sup><i>o</i></sup> = (1/2)[{–ln <i>f</i><sub><i>a</i><i>a</i><sup>[prime]</sup></sub> + <ln <i>f</i><sub><i>a</i><i>a</i><sup>[prime]</sup></sub>>} + {–ln <i>f</i><sub><i>a</i><sup>[prime]</sup><i>a</i></sub> + <ln <i>f</i><sub><i>a</i><sup>[prime]</sup><i>a</i></sub>>}],](024901_1-div2_files/024901_1m14.gif)
![<i>f</i><sub><i>a</i><i>a</i><sup>[prime]</sup></sub> [equivalent] <i>f</i><sub><i>a</i><i>a</i><sup>[prime]</sup></sub>(<i>theta</i>,<i>phi</i>,<i>Theta</i>,<i>Phi</i>,<i>Psi</i>),](024901_1-div2_files/024901_1m15.gif)
![<i>f</i><sub><i>a</i><sup>[prime]</sup><i>a</i></sub> [equivalent] <i>f</i><sub><i>a</i><sup>[prime]</sup><i>a</i></sub>(<i>theta</i><sup>[prime]</sup>,<i>phi</i><sup>[prime]</sup>,<i>Theta</i><sup>[prime]</sup>,<i>Phi</i><sup>[prime]</sup>,<i>Psi</i><sup>[prime]</sup>),](024901_1-div2_files/024901_1m16.gif)
where faa(,,,,) is a probability density function for a residue of type a at the orientation (,,,,) relative to the residue of type a; it satisfies
![[integral]<i>f</i><sub><i>a</i><i>a</i><sup>[prime]</sup></sub>(<i>theta</i>,<i>phi</i>,<i>Theta</i>,<i>Phi</i>,<i>Psi</i>)<i>d</i> cos <i>theta</i> <i>d</i> <i>phi</i> <i>d</i> cos <i>Theta</i> <i>d</i> <i>Phi</i> <i>d</i> <i>Psi</i> = 1.](024901_1-div2_files/024901_1m17.gif)
and faa:
![<i>Theta</i><sup>[prime]</sup> = –<i>Theta</i>, <i>Phi</i><sup>[prime]</sup> = –<i>Psi</i>, <i>Psi</i><sup>[prime]</sup> = –<i>Phi</i>.](024901_1-div2_files/024901_1m18.gif)
The relationship in respect to the polar angles (,) is not simple, but (,) can be uniquely calculated from (,,,,). Thus, in principle, faa and faa must be equal to each other:
![<i>f</i><sub><i>a</i><i>a</i><sup>[prime]</sup></sub>(<i>theta</i>,<i>phi</i>,<i>Theta</i>,<i>Phi</i>,<i>Psi</i>) = <i>f</i><sub><i>a</i><sup>[prime]</sup><i>a</i></sub>(<i>theta</i><sup>[prime]</sup>,<i>phi</i><sup>[prime]</sup>,<i>Theta</i><sup>[prime]</sup>,<i>Phi</i><sup>[prime]</sup>,<i>Psi</i><sup>[prime]</sup>).](024901_1-div2_files/024901_1m19.gif)
However, in the present statistical estimation of the probability density, the relationship above would be approximately satisfied. Therefore, the potential is evaluated in the form of Eq. (11).
![<–ln <i>f</i><sub><i>a</i><i>a</i><sup>[prime]</sup></sub>(<i>theta</i>,<i>phi</i>,<i>Theta</i>,<i>Phi</i>,<i>Psi</i>)> [equivalent] [integral] – <i>f</i><sub><i>a</i><i>a</i><sup>[prime]</sup></sub> ln <i>f</i><sub><i>a</i><i>a</i><sup>[prime]</sup></sub><i>d</i> cos <i>theta</i> <i>d</i> <i>phi</i> <i>d</i> cos <i>Theta</i> <i>d</i> <i>Phi</i> <i>d</i> <i>Psi</i>.](024901_1-div2_files/024901_1m20.gif)
Here it is important to note that this term represents a reference state such that the expected value of the orientational energy for each type of contacting residue pair in the native structures is equal to zero. Thus, this orientational potential represents simply the suitability of a relative orientation between contacting residues, but does not represent at all whether a contact between residues is favorable or not. The latter is supposed to be represented in the present scheme by the usual contact energy e. The reference distribution of residue-residue orientations for these orientational potentials is the uniform distribution, and not the overall distribution for all types of amino acid pairs employed by others.33,34,35 Therefore, for residue pairs whose distributions coincide with the overall distribution, the latter potentials give always no preference but the present potentials give a preference. This is a desirable behavior for orientational potentials, because such an overall distribution of residue-residue orientations would not be an intrinsic characteristic of non-native conformations but rather of native structures of proteins.
,,,,) variables.
![<i>f</i><sub><i>a</i><i>a</i><sup>[prime]</sup></sub>(<i>theta</i>,<i>phi</i>,<i>Theta</i>,<i>Phi</i>,<i>Psi</i>) = [summation]<sub><i>l</i><sub><i>p</i></sub> = 0</sub>[summation]<sub><i>m</i><sub><i>p</i></sub> = –<i>l</i><sub><i>p</i></sub></sub><sup><i>l</i><sub><i>p</i></sub></sup>[summation]<sub><i>l</i><sub><i>e</i></sub> = 0</sub>[summation]<sub><i>m</i><sub><i>e</i></sub> = –<i>l</i><sub><i>e</i></sub></sub><sup><i>l</i><sub><i>e</i></sub></sup>[summation]<sub><i>k</i><sub><i>e</i></sub></sub><i>c</i><sub><i>l</i><sub><i>p</i></sub><i>m</i><sub><i>p</i></sub><i>l</i><sub><i>e</i></sub><i>m</i><sub><i>e</i></sub><i>k</i><sub><i>e</i></sub></sub><sup><i>a</i><i>a</i><sup>[prime]</sup></sup><i>g</i><sub><i>l</i><sub><i>p</i></sub><i>m</i><sub><i>p</i></sub><i>l</i><sub><i>e</i></sub><i>m</i><sub><i>e</i></sub><i>k</i><sub><i>e</i></sub></sub>(<i>theta</i>,<i>phi</i>,<i>Theta</i>,<i>Phi</i>,<i>Psi</i>),](024901_1-div2_files/024901_1m21.gif)
g is represented as
![<i>g</i><sub><i>l</i><sub><i>p</i></sub><i>m</i><sub><i>p</i></sub><i>l</i><sub><i>e</i></sub><i>m</i><sub><i>e</i></sub><i>k</i><sub><i>e</i></sub></sub> [equivalent] <i>Y</i><sub><i>l</i><sub><i>p</i></sub></sub><sup><i>m</i><sub><i>p</i></sub></sup>(cos <i>theta</i>,<i>phi</i>)<i>Y</i><sub><i>l</i><sub><i>e</i></sub></sub><sup><i>m</i><sub><i>e</i></sub></sup>(cos <i>Theta</i>,<i>Phi</i>)<i>R</i><sub><i>k</i><sub><i>e</i></sub></sub>(<i>Psi</i>),](024901_1-div2_files/024901_1m22.gif)
![<i>Y</i><sub><i>l</i></sub><sup><i>m</i></sup>(cos <i>theta</i>,<i>phi</i>) = [(((2<i>l</i> + 1)(<i>l</i> – |<i>m</i>|)!)/(2(<i>l</i> + |<i>m</i>|)!))]<sup>1/2</sup><i>P</i><sub><i>l</i></sub><sup>|<i>m</i>|</sup>(cos <i>theta</i>)<i>R</i><sub><i>m</i></sub>(<i>phi</i>),](024901_1-div2_files/024901_1m23.gif)
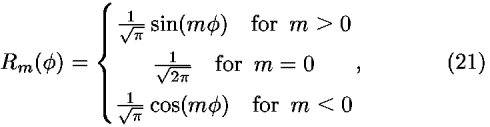
where Y is the normalized spherical harmonics function, P
is the normalized spherical harmonics function, P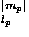 is the associated Legendre function; P
is the associated Legendre function; P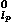 with mp = 0 is the Legendre polynomial. Then, the coefficients in the expansion of Eq. (18) can be calculated from the observed density distribution by
with mp = 0 is the Legendre polynomial. Then, the coefficients in the expansion of Eq. (18) can be calculated from the observed density distribution by
![<i>c</i><sub><i>l</i><sub><i>p</i></sub><i>m</i><sub><i>p</i></sub><i>l</i><sub><i>e</i></sub><i>m</i><sub><i>e</i></sub><i>k</i><sub><i>e</i></sub></sub><sup><i>a</i><i>a</i><sup>[prime]</sup></sup> = [integral]<i>f</i><sub><i>a</i><i>a</i><sup>[prime]</sup></sub><i>g</i><sub><i>l</i><sub><i>p</i></sub><i>m</i><sub><i>p</i></sub><i>l</i><sub><i>e</i></sub><i>m</i><sub><i>e</i></sub><i>k</i><sub><i>e</i></sub></sub><i>d</i> cos <i>theta</i> <i>d</i> <i>phi</i> <i>d</i> cos <i>Theta</i> <i>d</i> <i>Phi</i> <i>d</i> <i>Psi</i>.](024901_1-div2_files/024901_1m28.gif)
Thus, the coefficient of the first constant term in Eq. (18) that corresponds to the uniform distribution is obvious;
![<i>c</i><sub>00000</sub><sup><i>a</i><i>a</i><sup>[prime]</sup></sup> = (1/(2(2 <i>pi</i>)<sup>3/2</sup>)).](024901_1-div2_files/024901_1m29.gif)
function can be used,33 that is,
![<i>f</i><sub><i>a</i><i>a</i><sup>[prime]</sup></sub><sup><i>o</i><i>b</i><i>s</i></sup>(<i>theta</i>,<i>phi</i>,<i>Theta</i>,<i>Phi</i>,<i>Psi</i>) = (1/<i>N</i><sub><i>a</i><i>a</i><sup>[prime]</sup></sub>) [summation]<sub><i>µ</i> [is-an-element-of] {(<i>a</i><i>a</i><sup>[prime]</sup>)}</sub><i>w</i><sub><i>µ</i></sub><i>delta</i>(cos <i>theta</i> – cos <i>theta</i><sub><i>µ</i></sub>)<i>delta</i>(<i>phi</i> – <i>phi</i><sub><i>µ</i></sub>)<i>delta</i>(cos <i>Theta</i> – cos <i>Theta</i><sub><i>µ</i></sub>)<i>delta</i>(<i>Phi</i> – <i>Phi</i><sub><i>µ</i></sub>)<i>delta</i>(<i>Psi</i> – <i>Psi</i><sub><i>µ</i></sub>),](024901_1-div2_files/024901_1m30.gif)
![<i>N</i><sub><i>a</i><i>a</i><sup>[prime]</sup></sub> = [summation]<sub><i>µ</i> [is-an-element-of] {(<i>a</i><i>a</i><sup>[prime]</sup>)}</sub><i>w</i><sub><i>µ</i></sub>,](024901_1-div2_files/024901_1m31.gif)
and then, the expansion coefficients are calculated as
![<i>c</i><sub><i>l</i><sub><i>p</i></sub><i>m</i><sub><i>p</i></sub><i>l</i><sub><i>e</i></sub><i>m</i><sub><i>e</i></sub><i>k</i><sub><i>e</i></sub></sub><sup><i>a</i><i>a</i><sup>[prime]</sup></sup> = (1/<i>N</i><sub><i>a</i><i>a</i><sup>[prime]</sup></sub>) [summation]<sub><i>µ</i> [is-an-element-of] {(<i>a</i><i>a</i><sup>[prime]</sup>)}</sub><i>w</i><sub><i>µ</i></sub><i>g</i><sub><i>l</i><sub><i>p</i></sub><i>m</i><sub><i>p</i></sub><i>l</i><sub><i>e</i></sub><i>m</i><sub><i>e</i></sub><i>k</i><sub><i>e</i></sub></sub>(<i>theta</i><sub><i>µ</i></sub>,<i>phi</i><sub><i>µ</i></sub>,<i>Theta</i><sub><i>µ</i></sub>,<i>Phi</i><sub><i>µ</i></sub>,<i>Psi</i><sub><i>µ</i></sub>),](024901_1-div2_files/024901_1m32.gif)
where (µ,µ,µ,µ,µ) is a set of angles observed for the contact µ between residue types a and a, and wµ is a weight for this contact. The summations in the equations above are over all contacts of amino acid types a versus a. A contact between amino acid types a and a is counted as one half of a contact for a versus a and another half for a versus a; Naa + Naa is equal to the actual number of contacts between amino acid types a and a. Thus, a weight wµ is equal to 0.5wc, where wc is a sampling weight for each protein that is described in the section "Datasets of protein structures used." In Eq. (24), residues are regarded to be in contact if the geometric centers of side chains or C atoms for GLY are within 6.5 Ĺ.
![<i>c</i><sub><i>l</i><sub><i>p</i></sub><i>m</i><sub><i>p</i></sub><i>l</i><sub><i>e</i></sub><i>m</i><sub><i>e</i></sub><i>k</i><sub><i>e</i></sub></sub><sup><i>a</i><i>a</i><sup>[prime]</sup></sup> [approximate] (1/(1 + <i>beta</i><sub><i>l</i><sub><i>p</i></sub><i>m</i><sub><i>p</i></sub><i>l</i><sub><i>e</i></sub><i>m</i><sub><i>e</i></sub><i>k</i><sub><i>e</i></sub></sub><sup><i>a</i><i>a</i><sup>[prime]</sup></sup>)) [<i>beta</i><sub><i>l</i><sub><i>p</i></sub><i>m</i><sub><i>p</i></sub><i>l</i><sub><i>e</i></sub><i>m</i><sub><i>e</i></sub><i>k</i><sub><i>e</i></sub></sub><sup><i>a</i><i>a</i><sup>[prime]</sup></sup><i>c</i><sub><i>l</i><sub><i>p</i></sub><i>m</i><sub><i>p</i></sub><i>l</i><sub><i>e</i></sub><i>m</i><sub><i>e</i></sub><i>k</i><sub><i>e</i></sub></sub><sup><i>a</i><i>r</i></sup>+(1/<i>N</i><sub><i>a</i><i>a</i><sup>[prime]</sup></sub>) [summation]<sub><i>µ</i> [is-an-element-of] {(<i>a</i><i>a</i><sup>[prime]</sup>)}</sub><i>w</i><sub><i>µ</i></sub><i>g</i><sub><i>l</i><sub><i>p</i></sub><i>m</i><sub><i>p</i></sub><i>l</i><sub><i>e</i></sub><i>m</i><sub><i>e</i></sub><i>k</i><sub><i>e</i></sub></sub>(<i>theta</i><sub><i>µ</i></sub>,<i>phi</i><sub><i>µ</i></sub>,<i>Theta</i><sub><i>µ</i></sub>,<i>Phi</i><sub><i>µ</i></sub>,<i>Psi</i><sub><i>µ</i></sub>)],](024901_1-div2_files/024901_1m33.gif)
![<i>c</i><sub><i>l</i><sub><i>p</i></sub><i>m</i><sub><i>p</i></sub><i>l</i><sub><i>e</i></sub><i>m</i><sub><i>e</i></sub><i>k</i><sub><i>e</i></sub></sub><sup><i>a</i><i>r</i></sup> [approximate] (1/(1 + <i>beta</i><sub><i>l</i><sub><i>p</i></sub><i>m</i><sub><i>p</i></sub><i>l</i><sub><i>e</i></sub><i>m</i><sub><i>e</i></sub><i>k</i><sub><i>e</i></sub></sub><sup><i>a</i><i>r</i></sup>)) [<i>beta</i><sub><i>l</i><sub><i>p</i></sub><i>m</i><sub><i>p</i></sub><i>l</i><sub><i>e</i></sub><i>m</i><sub><i>e</i></sub><i>k</i><sub><i>e</i></sub></sub><sup><i>a</i><i>r</i></sup><i>c</i><sub><i>l</i><sub><i>p</i></sub><i>m</i><sub><i>p</i></sub><i>l</i><sub><i>e</i></sub><i>m</i><sub><i>e</i></sub><i>k</i><sub><i>e</i></sub></sub><sup><i>r</i><i>r</i></sup>+(1/<i>N</i><sub><i>a</i><i>r</i></sub>) [summation]<sub><i>µ</i> [is-an-element-of] {(<i>a</i><i>r</i>)}</sub><i>w</i><sub><i>µ</i></sub><i>g</i><sub><i>l</i><sub><i>p</i></sub><i>m</i><sub><i>p</i></sub><i>l</i><sub><i>e</i></sub><i>m</i><sub><i>e</i></sub><i>k</i><sub><i>e</i></sub></sub>(<i>theta</i><sub><i>µ</i></sub>,<i>phi</i><sub><i>µ</i></sub>,<i>Theta</i><sub><i>µ</i></sub>,<i>Phi</i><sub><i>µ</i></sub>,<i>Psi</i><sub><i>µ</i></sub>)],](024901_1-div2_files/024901_1m34.gif)
![<i>c</i><sub><i>l</i><sub><i>p</i></sub><i>m</i><sub><i>p</i></sub><i>l</i><sub><i>e</i></sub><i>m</i><sub><i>e</i></sub><i>k</i><sub><i>e</i></sub></sub><sup><i>r</i><i>a</i><sup>[prime]</sup></sup> [approximate] (1/(1 + <i>beta</i><sub><i>l</i><sub><i>p</i></sub><i>m</i><sub><i>p</i></sub><i>l</i><sub><i>e</i></sub><i>m</i><sub><i>e</i></sub><i>k</i><sub><i>e</i></sub></sub><sup><i>r</i><i>a</i><sup>[prime]</sup></sup>)) [<i>beta</i><sub><i>l</i><sub><i>p</i></sub><i>m</i><sub><i>p</i></sub><i>l</i><sub><i>e</i></sub><i>m</i><sub><i>e</i></sub><i>k</i><sub><i>e</i></sub></sub><sup><i>r</i><i>a</i><sup>[prime]</sup></sup><i>c</i><sub><i>l</i><sub><i>p</i></sub><i>m</i><sub><i>p</i></sub><i>l</i><sub><i>e</i></sub><i>m</i><sub><i>e</i></sub><i>k</i><sub><i>e</i></sub></sub><sup><i>r</i><i>r</i></sup>+(1/<i>N</i><sub><i>r</i><i>a</i><sup>[prime]</sup></sub>) [summation]<sub><i>µ</i> [is-an-element-of] {(<i>r</i><i>a</i><sup>[prime]</sup>)}</sub><i>w</i><sub><i>µ</i></sub><i>g</i><sub><i>l</i><sub><i>p</i></sub><i>m</i><sub><i>p</i></sub><i>l</i><sub><i>e</i></sub><i>m</i><sub><i>e</i></sub><i>k</i><sub><i>e</i></sub></sub>(<i>theta</i><sub><i>µ</i></sub>,<i>phi</i><sub><i>µ</i></sub>,<i>Theta</i><sub><i>µ</i></sub>,<i>Phi</i><sub><i>µ</i></sub>,<i>Psi</i><sub><i>µ</i></sub>)],](024901_1-div2_files/024901_1m35.gif)
![<i>c</i><sub><i>l</i><sub><i>p</i></sub><i>m</i><sub><i>p</i></sub><i>l</i><sub><i>e</i></sub><i>m</i><sub><i>e</i></sub><i>k</i><sub><i>e</i></sub></sub><sup><i>r</i><i>r</i></sup> [approximate] (1/(1 + <i>beta</i><sub>00000</sub><sup><i>r</i><i>r</i></sup>)) [<i>beta</i><sub>00000</sub><sup><i>r</i><i>r</i></sup><i>c</i><sub>00000</sub><sup><i>r</i><i>r</i></sup><i>delta</i><sub>0<i>l</i><sub><i>p</i></sub></sub><i>delta</i><sub>0<i>m</i><sub><i>p</i></sub></sub><i>delta</i><sub>0<i>l</i><sub><i>e</i></sub></sub><i>delta</i><sub>0<i>m</i><sub><i>p</i></sub></sub><i>delta</i><sub>0<i>k</i><sub><i>e</i></sub></sub>+(1/<i>N</i><sub><i>r</i><i>r</i></sub>) [summation]<sub><i>µ</i> [is-an-element-of] {(<i>r</i><i>r</i>)}</sub><i>w</i><sub><i>µ</i></sub><i>g</i><sub><i>l</i><sub><i>p</i></sub><i>m</i><sub><i>p</i></sub><i>l</i><sub><i>e</i></sub><i>m</i><sub><i>e</i></sub><i>k</i><sub><i>e</i></sub></sub>(<i>theta</i><sub><i>µ</i></sub>,<i>phi</i><sub><i>µ</i></sub>,<i>Theta</i><sub><i>µ</i></sub>,<i>Phi</i><sub><i>µ</i></sub>,<i>Psi</i><sub><i>µ</i></sub>)],](024901_1-div2_files/024901_1m36.gif)
where 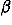
![<sub><i>l</i><sub><i>p</i></sub><i>m</i><sub><i>p</i></sub><i>l</i><sub><i>e</i></sub><i>m</i><sub><i>e</i></sub><i>k</i><sub><i>e</i></sub></sub><sup><i>a</i><i>a</i><sup>[prime]</sup></sup>](024901_1-div2_files/024901_1m37.gif) is taken to be
is taken to be
![<i>beta</i><sub><i>l</i><sub><i>p</i></sub><i>m</i><sub><i>p</i></sub><i>l</i><sub><i>e</i></sub><i>m</i><sub><i>e</i></sub><i>k</i><sub><i>e</i></sub></sub><sup><i>a</i><i>a</i><sup>[prime]</sup></sup> [equivalent] ((<i>beta</i> <i>O</i><sub><i>l</i><sub><i>p</i></sub><i>m</i><sub><i>p</i></sub><i>l</i><sub><i>e</i></sub><i>m</i><sub><i>e</i></sub><i>k</i><sub><i>e</i></sub></sub>)/<i>N</i><sub><i>a</i><i>a</i><sup>[prime]</sup></sub>),](024901_1-div2_files/024901_1m38.gif)
![<i>O</i><sub><i>l</i><sub><i>p</i></sub><i>m</i><sub><i>p</i></sub><i>l</i><sub><i>e</i></sub><i>m</i><sub><i>e</i></sub><i>k</i><sub><i>e</i></sub></sub> [equivalent] [the number of frequency modes lower than or equal to (<i>l</i><sub><i>p</i></sub>,<i>m</i><sub><i>p</i></sub>,<i>l</i><sub><i>e</i></sub>,<i>m</i><sub><i>e</i></sub>,<i>k</i><sub><i>e</i></sub>)] = (<i>l</i><sub><i>p</i></sub><sup>2</sup> + 2|<i>m</i><sub><i>p</i></sub>| + 1)(<i>l</i><sub><i>e</i></sub><sup>2</sup> + 2|<i>m</i><sub><i>e</i></sub>| + 1)(2|<i>k</i><sub><i>e</i></sub>| + 1),](024901_1-div2_files/024901_1m39.gif)
in order to reduce statistical errors resulting from the small size of samples; in Eq. (31) is a parameter to be optimized. Equation (31) means that more samples are required to determine higher frequency modes. In Eq. (27), the first term becomes more effective than the second term in the limit of small numbers of Naa, and inversely the second term becomes more effective than the first term in the limit of large numbers of Naa.
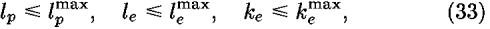
and

where Ocutoff is a cutoff value for expansion terms.
![<i>f</i><sub><i>a</i><i>a</i><sup>[prime]</sup></sub>(<i>theta</i>,<i>phi</i>,<i>Theta</i>,<i>Phi</i>,<i>Psi</i>) [approximate] [summation]<sub><i>l</i><sub><i>p</i></sub> = 0</sub><sup><i>l</i><sub><i>p</i></sub><sup>max</sup></sup>[summation]<sub><i>m</i><sub><i>p</i></sub> = –<i>l</i><sub><i>p</i></sub></sub><sup><i>l</i><sub><i>p</i></sub></sup>[summation]<sub><i>l</i><sub><i>e</i></sub> = 0</sub><sup><i>l</i><sub><i>e</i></sub><sup>max</sup></sup>[summation]<sub><i>m</i><sub><i>e</i></sub> = –<i>l</i><sub><i>e</i></sub></sub><sup><i>l</i><sub><i>e</i></sub></sup>[summation]<sub><i>k</i><sub><i>e</i></sub></sub><sup><i>k</i><sub><i>e</i></sub><sup>max</sup></sup><i>H</i>(<i>O</i><sub>cutoff</sub> – <i>O</i><sub><i>l</i><sub><i>p</i></sub><i>m</i><sub><i>p</i></sub><i>l</i><sub><i>e</i></sub><i>m</i><sub><i>e</i></sub><i>k</i><sub><i>e</i></sub></sub>)<i>H</i>(|<i>c</i><sub><i>l</i><sub><i>p</i></sub><i>m</i><sub><i>p</i></sub><i>l</i><sub><i>e</i></sub><i>m</i><sub><i>e</i></sub><i>k</i><sub><i>e</i></sub></sub><sup><i>a</i><i>a</i><sup>[prime]</sup></sup>| – <i>c</i><sub>cutoff</sub><i>c</i><sub>00000</sub><sup><i>a</i><i>a</i><sup>[prime]</sup></sup>)<i>c</i><sub><i>l</i><sub><i>p</i></sub><i>m</i><sub><i>p</i></sub><i>l</i><sub><i>e</i></sub><i>m</i><sub><i>e</i></sub><i>k</i><sub><i>e</i></sub></sub><sup><i>a</i><i>a</i><sup>[prime]</sup></sup><i>g</i><sub><i>l</i><sub><i>p</i></sub><i>m</i><sub><i>p</i></sub><i>l</i><sub><i>e</i></sub><i>m</i><sub><i>e</i></sub><i>k</i><sub><i>e</i></sub></sub>(<i>theta</i>,<i>phi</i>,<i>Theta</i>,<i>Phi</i>,<i>Psi</i>),](024901_1-div2_files/024901_1m42.gif)
where H is the Heaviside step function which takes a value of one for zero and positive values of the argument and is otherwise zero. Finally the estimate of the probability density faa(,,,,) is cut off at sufficiently low and high values in such a way that its logarithm takes a value within an appropriate range; for example, –7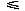 –ln faa(,,,,) + ln(c
–ln faa(,,,,) + ln(c![<sub>00000</sub><sup><i>a</i><i>a</i><sup>[prime]</sup></sup>](024901_1-div2_files/024901_1m43.gif) g00000)1.
g00000)1.
![<–ln <i>f</i><sub><i>a</i><i>a</i><sup>[prime]</sup></sub>(<i>theta</i>,<i>phi</i>,<i>Theta</i>,<i>Phi</i>,<i>Psi</i>)> [approximate] ((–1)/<i>N</i><sub><i>a</i><i>a</i><sup>[prime]</sup></sub>) [summation]<sub><i>µ</i> [is-an-element-of] {(<i>a</i><i>a</i><sup>[prime]</sup>)}</sub><i>w</i><sub><i>µ</i></sub> ln <i>f</i><sub><i>a</i><i>a</i><sup>[prime]</sup></sub>(<i>theta</i><sub><i>µ</i></sub>,<i>phi</i><sub><i>µ</i></sub>,<i>Theta</i><sub><i>µ</i></sub>,<i>Phi</i><sub><i>µ</i></sub>,<i>Psi</i><sub><i>µ</i></sub>).](024901_1-div2_files/024901_1m44.gif)
 , and a repulsive packing potential e
, and a repulsive packing potential e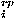 ,
,
![<i>E</i><sup><i>r</i></sup> = [summation]<sub><i>i</i></sub>{(1/2) [summation]<sub><i>j</i></sub><i>e</i><sup><i>h</i><i>c</i></sup>(<b>r</b><sub><i>i</i></sub>,<b>r</b><sub><i>j</i></sub>) + <i>e</i><sub><i>i</i><i>j</i></sub><sup><i>r</i><i>c</i></sup> + <i>e</i><sub><i>i</i></sub><sup><i>r</i><i>p</i></sup>},](024901_1-div2_files/024901_1m47.gif)
![<i>e</i><sup><i>h</i><i>c</i></sup>(<b>r</b><sub><i>i</i></sub>,<b>r</b><sub><i>j</i></sub>) [equivalent] 10<i>S</i><sub><i>w</i></sub>(|<b>r</b><sub><i>i</i></sub> – <b>r</b><sub><i>j</i></sub>|,2.2,2.6),](024901_1-div2_files/024901_1m48.gif)
![<i>e</i><sub><i>i</i><i>j</i></sub><sup><i>r</i><i>e</i></sup> = <i>H</i>(<i>n</i><sub><i>i</i></sub><sup><i>c</i></sup> – <i>q</i><sub><i>a</i><sub><i>i</i></sub></sub><sup><i>c</i></sup>)[((<i>q</i><sub><i>a</i><sub><i>i</i></sub></sub><sup><i>c</i></sup>/<i>n</i><sub><i>i</i></sub><sup><i>c</i></sup>) – 1)<i>e</i><sup><i>c</i></sup>(<b>r</b><sub><i>i</i></sub>,<b>r</b><sub><i>j</i></sub>)],](024901_1-div2_files/024901_1m49.gif)
![<i>e</i><sub><i>i</i></sub><sup><i>r</i><i>p</i></sup> = <i>H</i>(<i>n</i><sub><i>i</i></sub><sup><i>c</i></sup> – <i>q</i><sub><i>a</i><sub><i>i</i></sub></sub><sup><i>c</i></sup>)[–ln(((<i>N</i>(<i>a</i><sub><i>i</i></sub>,<i>n</i><sub><i>i</i></sub><sup><i>c</i></sup>) + <i>epsilon</i>)/(<i>N</i>(<i>a</i><sub><i>i</i></sub>,<i>q</i><sub><i>a</i><sub><i>i</i></sub></sub><sup><i>c</i></sup>) + <i>epsilon</i>)))],](024901_1-div2_files/024901_1m50.gif)
![<i>n</i><sub><i>i</i></sub><sup><i>c</i></sup> = [summation]<sub><i>j</i></sub> <i>Delta</i><sup><i>c</i></sup>(<b>r</b><sub><i>i</i></sub>,<b>r</b><sub><i>j</i></sub>),](024901_1-div2_files/024901_1m51.gif)
where Sw is defined by Eq. (7). The repulsive packing potentials e for the 20 types of residues are estimated from the observed distributions of the numbers of contacting residues in dense regions of protein structures by assuming a Boltzmann distribution.3 N(ai,n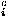 ) is the observed number of residues of type ai that are surrounded by n residues in the database of protein structures. q
) is the observed number of residues of type ai that are surrounded by n residues in the database of protein structures. q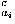 is a coordination number, which is defined as the maximum feasible number of contacting residues around a residue, for the amino acid of type ai.
is a coordination number, which is defined as the maximum feasible number of contacting residues around a residue, for the amino acid of type ai.  in Eq. (40) is a small value (= 10–6) that is added to avoid the divergence of the logarithm function. The observed distribution N(ai,n) used here is one44 compiled from 2129 protein species representatives of the SCOP48 Release 1.53 with our sampling method.3
in Eq. (40) is a small value (= 10–6) that is added to avoid the divergence of the logarithm function. The observed distribution N(ai,n) used here is one44 compiled from 2129 protein species representatives of the SCOP48 Release 1.53 with our sampling method.3
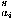 (i,
(i,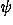 i) over all residues:
i) over all residues:
![<i>E</i><sup><i>s</i></sup> = [summation]<sub><i>i</i></sub><i>e</i><sub><i>a</i><sub><i>i</i></sub></sub><sup><i>s</i></sup>(<i>phi</i><sub><i>i</i></sub>,<i>psi</i><sub><i>i</i></sub>).](024901_1-div2_files/024901_1m55.gif)
For this secondary structure potential, a 10° mesh over (,) space is used to count frequencies of amino acids observed in protein native structures, and this intraresidue potential e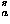 for each amino acid type a is evaluated as
for each amino acid type a is evaluated as
![<i>e</i><sub><i>a</i></sub><sup><i>s</i></sup>(<i>phi</i>,<i>psi</i>) [equivalent] –ln(<i>N</i><sub><i>a</i></sub>(<i>phi</i>,<i>psi</i>)/<i>N</i><sub><i>a</i></sub>) + <ln(<i>N</i><sub><i>a</i></sub>(<i>phi</i>,<i>psi</i>)/<i>N</i><sub><i>a</i></sub>)>,](024901_1-div2_files/024901_1m57.gif)
![<–ln(<i>N</i><sub><i>a</i></sub>(<i>phi</i>,<i>psi</i>)/<i>N</i><sub><i>a</i></sub>)> = ((–1)/<i>N</i><sub><i>a</i></sub>) [summation]<sub>(<i>phi</i>,<i>psi</i>)</sub><i>N</i><sub><i>a</i></sub>(<i>phi</i>,<i>psi</i>)ln(<i>N</i><sub><i>a</i></sub>(<i>phi</i>,<i>psi</i>)/<i>N</i><sub><i>a</i></sub>),](024901_1-div2_files/024901_1m58.gif)
where Na(,) is the number of amino acids of type a at (,) observed in protein native structures, and Na is their sum over the entire (,) space, that is, the number of amino acids of type a. The second term is a constant term that corresponds to a reference energy, so that the (,) energy expected for each type of residue in the native structures is equal to zero.
,) used here is one44 compiled from 2129 protein species representatives of the SCOP48 Release 1.53 with the sampling method3 used to reduce the weights of contributions of structures having high sequence identity.
, all , /, + , and multidomain proteins. Classes of membrane and cell surface proteins, small proteins, peptides, and designed proteins are not used. Proteins whose structures50 were determined by NMR or having stated resolutions worse than 2.5 Ĺ are removed to assure that the quality of proteins used is high. Also, proteins whose coordinate sets consist either of only C atoms, or include many unknown residues, or lack many atoms or residues, are removed. In addition, proteins shorter than 50 residues are also removed. As a result, the set of species representatives includes 4435 protein domains; this dataset is named here as dataset A.