The Journal of Chemical Physics, 08 January 2005
J. Chem. Phys. 122, 024901 (2005) (18 pages)
©2005 American Institute of Physics. All rights reserved.
Previous section: II. METHODS
Next section: IV. DISCUSSION
Title Page
III. RESULTS
A. Local coordinate system affixed to each residue
In order to describe the relative directional and rotational positions of contacting residues, a local coordinate system defined as in Fig. 1 is affixed to each residue. Here the local coordinate system is defined for fold recognition, based only on the main chain atoms of N, C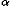 , and C
, and C![[prime]](024901_1-div3_files/prime-script.gif) to represent the orientational relationship between the main chains of contacting residues rather than representing33,34,35 those relationships between the side chains. The origin O of the local coordinate system is located at the C position of each residue. The Y and Z axes are ones formed by the vector product and the sum of the unit vectors from N to C and from C to C, respectively. That is, the Y and Z axes are taken to be perpendicular to and in the plane of the three atoms N, C, and C, respectively. These form a right-handed coordinate system. There are two degrees of directional freedom and three degrees of rotational freedom in the relative orientation of one residue to another in contacting residue pairs. The relative direction and rotation of one residue to another in contacting residues are represented by polar angles (
to represent the orientational relationship between the main chains of contacting residues rather than representing33,34,35 those relationships between the side chains. The origin O of the local coordinate system is located at the C position of each residue. The Y and Z axes are ones formed by the vector product and the sum of the unit vectors from N to C and from C to C, respectively. That is, the Y and Z axes are taken to be perpendicular to and in the plane of the three atoms N, C, and C, respectively. These form a right-handed coordinate system. There are two degrees of directional freedom and three degrees of rotational freedom in the relative orientation of one residue to another in contacting residue pairs. The relative direction and rotation of one residue to another in contacting residues are represented by polar angles (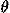 ,
,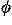 ) and Euler angles (
) and Euler angles (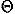 ,
,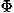 ,
,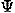 ), respectively.
), respectively.
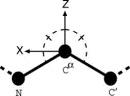 Figure 1.
Figure 1. B. Orientational distributions of contacting residues
Release 1.61 of the SCOP database48 for classification of protein folds has been used to choose representatives for different protein folds. In the 4435 chosen representative proteins, which correspond to the 3522 effective number of sequences, the 1 467 302 effective number of residue-residue contacts are observed and used here to evaluate the statistical distribution of relative residue-residue orientations for each type of residue pair. The orientational distributions are evaluated in the multimeric state of a whole protein structure for each protein domain.
As described in the Methods section, the sample size limits the frequencies of modes whose expansion coefficients can be reliably estimated. Here, values in the range 4–14 are used for l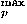 , l
, l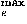 , and k that are the maximum values of lp, le, and ke which are the highest frequency modes to be estimated. However, even though each of (lp, mp, le, me, ke) is sufficiently small, their combinations may correspond to high frequency modes. The number of modes lower than or equal to (lp, mp, le, me, ke), Olpmplemeke defined by Eq. (32), is used as a one-dimensional projection of (lp, mp, le, me, ke) on a frequency axis. To remove high frequency modes, only frequency modes less than and equal to Ocutoff are utilized. In addition, only significant terms in the expansion of Eq. (35) whose coefficients take larger absolute values than the value of a cutoff, ccutoffc
, and k that are the maximum values of lp, le, and ke which are the highest frequency modes to be estimated. However, even though each of (lp, mp, le, me, ke) is sufficiently small, their combinations may correspond to high frequency modes. The number of modes lower than or equal to (lp, mp, le, me, ke), Olpmplemeke defined by Eq. (32), is used as a one-dimensional projection of (lp, mp, le, me, ke) on a frequency axis. To remove high frequency modes, only frequency modes less than and equal to Ocutoff are utilized. In addition, only significant terms in the expansion of Eq. (35) whose coefficients take larger absolute values than the value of a cutoff, ccutoffc![<sub>00000</sub><sup><i>a</i><i>a</i><sup>[prime]</sup></sup>](024901_1-div3_files/024901_1m43.gif) , are used to estimate the distributions of relative residue-residue orientations.
, are used to estimate the distributions of relative residue-residue orientations.
Deviations from the uniform distribution in the estimated orientational distributions can be measured by reductions in orientational entropy. In the case of the uniform distribution, the orientational entropy defined by Eq. (17) is equal to –ln(cg00000) = 6.900 in kB units; kB is the Boltzmann constant. The estimate of orientational entropy for each type of residue pair and the number of significant terms required for the estimation depends on the resolution of the potentials, that is, the values of l, l, and k, and also the cutoff parameters of Ocutoff and ccutoff, and 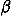 for the correction for a small sample size. Orientational entropies estimated with various values of the parameters are shown in Fig. 2, and the numbers of significant terms required are plotted in Fig. 3. Orientational entropies and the numbers of significant terms averaged with a weight of the number of contacts over all residue pairs are plotted against the cutoff value of the coefficient for expansion terms, ccutoff. Triples of digits near curves in the figure indicate the values of (l, l, and k). The entropy reduction is large when the resolution of the potential increases. The estimate of orientational entropy with l = l = k = 4,5,6 almost converges at the cutoff value, ccutoff = 0.025. The number of significant terms decreases almost exponentially with the cutoff value, ccutoff; see Fig. 3. The number of significant terms required for each type of residue pair is related to the orientational entropy for the residue pair. Figure 4 shows the correlation between the orientational entropies and the number of significant terms. As expected, many significant terms tend to be required for residue pairs whose orientational entropies are large. The frequency distribution of the number of significant terms for the 210 types of residue pairs is shown in Fig. 5, indicating that the orientational distribution strongly depends on the type of residue pair.
for the correction for a small sample size. Orientational entropies estimated with various values of the parameters are shown in Fig. 2, and the numbers of significant terms required are plotted in Fig. 3. Orientational entropies and the numbers of significant terms averaged with a weight of the number of contacts over all residue pairs are plotted against the cutoff value of the coefficient for expansion terms, ccutoff. Triples of digits near curves in the figure indicate the values of (l, l, and k). The entropy reduction is large when the resolution of the potential increases. The estimate of orientational entropy with l = l = k = 4,5,6 almost converges at the cutoff value, ccutoff = 0.025. The number of significant terms decreases almost exponentially with the cutoff value, ccutoff; see Fig. 3. The number of significant terms required for each type of residue pair is related to the orientational entropy for the residue pair. Figure 4 shows the correlation between the orientational entropies and the number of significant terms. As expected, many significant terms tend to be required for residue pairs whose orientational entropies are large. The frequency distribution of the number of significant terms for the 210 types of residue pairs is shown in Fig. 5, indicating that the orientational distribution strongly depends on the type of residue pair.
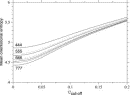 Figure 2.
Figure 2. 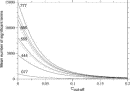 Figure 3.
Figure 3. 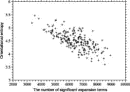 Figure 4.
Figure 4. 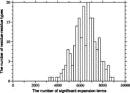 Figure 5.
Figure 5. The orientational entropies  –ln faa
–ln faa for each type of residue pair are listed in Table I. Residue type "r" in Table I means any type of residue. As already noted in the Methods section, in principle this matrix is symmetrical. The table shows that the matrix is almost symmetrical, indicating the good quality of their statistical estimates. These values in this table are calculated with l = l = k = 6, Ocutoff = O33333 = 1792, = 0.2, and ccutoff = 0.025.
for each type of residue pair are listed in Table I. Residue type "r" in Table I means any type of residue. As already noted in the Methods section, in principle this matrix is symmetrical. The table shows that the matrix is almost symmetrical, indicating the good quality of their statistical estimates. These values in this table are calculated with l = l = k = 6, Ocutoff = O33333 = 1792, = 0.2, and ccutoff = 0.025.
Orientational entropies for residue pairs with GLY appear to be relatively large. Also orientational entropies for residue pairs with PRO tend to be larger than those for others but smaller than those for residue pairs with GLY. Residue pairs TRP-CYS/CYS-TRP have the smallest orientational entropies. Orientational entropies for residue pairs with CYS and GLU are relatively small. As expected, CYS-CYS, GLU-GLU, GLU-ASP/ASP-GLU, and LYS-LYS have relatively small orientational entropies, probably because of S–S bond interactions and charge-charge interactions.
C. Distributions of residue orientations depend significantly on Euler angles
It is interesting to see how much the entropy reductions originate either from polar angle dependences or Euler angle dependences only, and from cross correlations between them; the orientational entropy is defined by Eq. (17) and estimated by Eq. (36).
In Fig. 6, the broken line shows the maximum value of orientational entropy which each type of amino acid pair can take; it is equal to –ln(cg00000) = 6.900 for the uniform distribution. The abscissa indicates the amino acid pair identification number; amino acid types are numbered in the order of amino acids written along the abscissa. Thus, the amino acid pair identification number one means a CYS-CYS pair and 400 means a PRO-PRO pair. The lowest solid line is for a distribution estimated with l = l = k = 6. The highest solid line shows the orientational entropies estimated with l = 6, l = k = 0, and therefore the contribution to the total entropies from polar angle dependences. The middle line shows the orientational entropies estimated by subtracting the entropy, 6.900, for the uniform distribution from the sum of entropies estimated with l = 6, l = k = 0, and with l = 0, l = k = 6. In other words, the difference between the highest solid line and the middle line shows contributions to the total entropies from Euler angle dependences. The difference between the middle and lowest solid lines corresponds to contributions from the cross correlation between polar angle and Euler angle dependences. Cutoff values for significant terms in the expansion are Ocutoff = 1792 and ccutoff = 0.025. The parameter for the correction for a small sample size is = 0.2.
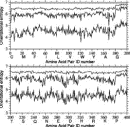 Figure 6.
Figure 6. These results clearly indicate that only small amounts of entropy reduction originate purely from polar angle dependences, and that the distribution of residue orientations has significantly large correlations between polar and Euler angles. Also, the fact that the lowest solid line is more jagged than the upper lines indicates that the distributions as a function of polar and Euler angles, can reflect more differences among the types of residue pairs than the others. Thus, the discriminations of native structures from non-native folds is expected to be improved by taking account of Euler angle dependencies in the distributions of residue-residue orientations.
D. Recognition power for native structures
We have evaluated the recognition power of the orientational potentials for native structures using independently constructed decoy sets, which are maintained at "http://dd.stanford.edu" as the database "Decoys'R'Us." 39 Here, the group of decoy sets named "multiple" are employed. This group of decoy sets consists of the following ten families of decoy sets classified by methods used to generate decoys. Each decoy set provides multiple non-native structures as well as the native structure.
(1) The "4state_reduced" family containing decoy sets for seven small proteins. C positions for these decoys were generated by exhaustively enumerating ten selectively chosen residues in each protein using a four-state off-lattice model.36
(2) The "fisa" family containing decoy sets for four 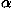 helical proteins. The main chains for these decoys were generated using a fragment insertion simulated annealing procedure to assemble nativelike structures from fragments of unrelated protein structures with similar local sequences using Bayesian scoring functions.37
helical proteins. The main chains for these decoys were generated using a fragment insertion simulated annealing procedure to assemble nativelike structures from fragments of unrelated protein structures with similar local sequences using Bayesian scoring functions.37
(3) The "fisa_casp3" containing decoy sets for proteins predicted by the Baker group for CASP3. The same method as for the fisa set was used to generate the main chains and side chains for these decoys.
(4) The "hg_structal" family containing decoy sets for 29 globins. Each decoy has been built by comparative modeling using 29 other globins as templates with the program "segmod." 51
(5) The "lattice_ssfit" family containing decoy sets for eight small proteins generated by ab initio methods.38
(6) The local minima decoy set family ("lmds") which containing decoy sets derived from the experimental secondary structures of ten small proteins belonging to diverse structural classes. Each decoy is at a local minimum of an energy function.
(7) The second version, "lmds_v2," of the local minima decoy set family, lmds.
(8) The "semfold" family containing decoy sets for six proteins.
(9) The "ig_structal" family containing decoy sets for 61 immunoglobulin domains. Each decoy has been built by comparative modeling using all the other immunoglobulins as templates with the program segmod.51
(10) The "ig_structal_hires" family that is a high resolution subset of ig_structal, and contains decoy sets for 20 immunoglobulins. The resolution range is for this set is 1.7–2.2 Ĺ compared to the range of 1.7–3.1 Ĺ for the full 61 set.
In the following, these families of decoy sets are categorized into two classes one of which consists of only the last two families above, i.e., the decoy set group of immunoglobulin domains that are single chains of a multimer, and the other which contains the rest of the decoy families above and is called the decoy set group of monomeric proteins; although hg_structal contains decoy sets for some hemoglobins which are tetrameric proteins, and the fragment B of protein A, which is in a complex with immunoglobulin Fc, is also contained as the decoy set 1FC2 in the decoy set families fisa, lmds, and lmds_v2. This classification that depends on whether decoys are a single chain of a multimer is based on the fact that the true ground state of those multimeric proteins requires all of the chains to be present; it is true especially for contact energies, although it is not expected for the orientational energies developed here or short-range potentials such as the secondary structure potentials. The decoy set group of monomeric proteins consists of 79 decoy sets, and the decoy set group of immunoglobulin domains consists of 81 decoy sets.
In the evaluation of the recognition performance of potential functions for the native structures, proteins contained in the decoy sets have been removed from a dataset of proteins from which the orientational potentials are compiled; that is, the dataset B is used.
E. Evaluation of the performance of potential functions in fold recognition
The performance of potential functions in fold recognition is evaluated for each decoy set by the rank, the logarithm of rank probability, and the Z score of the native fold in the energy scale, and by those of the lowest energy fold in the root mean square deviation (RMSD) scale. RMSD means the least root mean square deviation between C atoms in overlaps between the native structure and decoys. The rank probabilities, Pe in the energy scale and Pr in the RMSD scale, are defined as
![<i>P</i><sub><i>e</i></sub> [equivalent] the rank of the native fold in an energyscale/the number of decoys,](024901_1-div3_files/024901_1m61.gif)
![<i>P</i><sub><i>r</i></sub> [equivalent] the rank of the lowest energy fold in theRMSD scale/the number of decoys,](024901_1-div3_files/024901_1m62.gif)
The Z scores Ze in the energy scale and Zrmsd in the RMSD scale are defined as
![<i>Z</i><sub><i>e</i></sub> [equivalent] ((<i>E</i><sub>native</sub> – overline(<i>E</i><sub>decoy</sub>))/(<i>sigma</i><sub><i>E</i></sub>)),](024901_1-div3_files/024901_1m63.gif)
![<i>Z</i><sub><i>r</i></sub> [equivalent] <i>Z</i><sub>rmsd</sub> [equivalent] ((RMSD<sub>lowest</sub> – overline(RMSD<sub>decoy</sub>))/(<i>sigma</i><sub>rmsd</sub>)),](024901_1-div3_files/024901_1m64.gif)
where 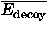 and
and 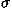 E are the mean and the standard deviation of energies of decoys, and
E are the mean and the standard deviation of energies of decoys, and 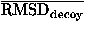 and rmsd are the mean and the standard deviation of RMSD of decoys. RMSDlowest is the RMSD of the lowest energy fold.
and rmsd are the mean and the standard deviation of RMSD of decoys. RMSDlowest is the RMSD of the lowest energy fold.
The correlation coefficient R of rank order between the energies and RMSDs of decoys is also listed in some tables, because it was used in Ref. 25.
F. How important are the Euler angle dependencies of relative residue orientations for fold recognition?
First, we examine how the discrimination power is improved by taking account of the Euler angle dependencies of relative orientations between residues. In the case of l = k = 0, Euler angle dependencies are completely ignored. Thus, the comparisons of the performances of discrimination between the cases of l = k = 0 and l,k![[not-equal]](024901_1-div3_files/ne.gif) 0 indicate how important the Euler angle dependencies of relative residue orientations are in fold recognition. In Tables II and III, the performances of discrimination are compared among some combinations of parameters l and l for both the decoy set groups of monomeric proteins and immunoglobulin domains; k was taken to be equal to l. The full lists of these tables are provided in the auxiliary material.52 Here, the potentials consist of the orientational potential eo only. In these tables, the performances of discrimination are evaluated by the number of decoy sets (no. of tops) in which the native structure is the lowest energy fold, and also the averages over the decoy sets of the logarithms of rank probabilities Pe in the energy scale and Pr in the RMSD scale, and the mean Z scores Ze of the native folds in the energy scale.
0 indicate how important the Euler angle dependencies of relative residue orientations are in fold recognition. In Tables II and III, the performances of discrimination are compared among some combinations of parameters l and l for both the decoy set groups of monomeric proteins and immunoglobulin domains; k was taken to be equal to l. The full lists of these tables are provided in the auxiliary material.52 Here, the potentials consist of the orientational potential eo only. In these tables, the performances of discrimination are evaluated by the number of decoy sets (no. of tops) in which the native structure is the lowest energy fold, and also the averages over the decoy sets of the logarithms of rank probabilities Pe in the energy scale and Pr in the RMSD scale, and the mean Z scores Ze of the native folds in the energy scale.
Table II(a) shows the dependencies of the recognition power on the resolution in polar angles; note that Euler angle dependencies are completely ignored with l = k = 0. Both the monomeric protein decoy set group and immunoglobulin decoy set group show similar characteristics; when the resolution, that is, the value of l increases up to 7, the number of top ranks tends to increase and the means of the log rank probabilities, 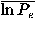 in the energy scale and
in the energy scale and 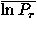 in the RMSD scale, tend to be improved with more negative values. The potentials with 7<l<14 appear to yield worse results than that of l = 7. At l = 14, the orientational potential shows a similar performance to that for l = 7. These results indicate that the improvement in the performance of fold recognition is not monotonic with the number of expansion terms, and also that there may be an intrinsic periodicity in the polar-angle distribution of residue-residue orientations.
in the RMSD scale, tend to be improved with more negative values. The potentials with 7<l<14 appear to yield worse results than that of l = 7. At l = 14, the orientational potential shows a similar performance to that for l = 7. These results indicate that the improvement in the performance of fold recognition is not monotonic with the number of expansion terms, and also that there may be an intrinsic periodicity in the polar-angle distribution of residue-residue orientations.
Similar performance is obtained for both the decoy set group by using the Euler angle distributions of residue-residue orientations. The dependencies of the recognition power on the resolution in Euler angles are shown in Table II(b). For this table, l = 0 is used, so that polar-angle dependencies are completely ignored. The best result in the cases of 4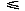 l = k7 is obtained in the case of the highest resolution, l = 0,l = k = 7. In comparison with the results of l = 7,l = k = 0, some improvement is clearly observed for the immunoglobulin decoy set group, although the performance of z score Ze is slightly worse for the monomeric protein decoy set group. The native structures of immunoglobulin domains consist mainly of sheets. Hydrogen bonds between strands are essential to maintain sheets. In addition to hydrogen bonds, residue-residue packing between a sheet and other parts may require relatively stringent orientations between residues, especially for Euler angles.
l = k7 is obtained in the case of the highest resolution, l = 0,l = k = 7. In comparison with the results of l = 7,l = k = 0, some improvement is clearly observed for the immunoglobulin decoy set group, although the performance of z score Ze is slightly worse for the monomeric protein decoy set group. The native structures of immunoglobulin domains consist mainly of sheets. Hydrogen bonds between strands are essential to maintain sheets. In addition to hydrogen bonds, residue-residue packing between a sheet and other parts may require relatively stringent orientations between residues, especially for Euler angles.
To improve the performance, correlations between polar and Euler angle dependencies must be taken into account. Table III shows the improvements in recognition performance obtained by taking account of the correlations between polar and Euler angle dependencies. Table III(a) indicates that the recognition performance is improved about 10% to 30% for both of the decoy set groups with increase of resolution, but has a limitation around l = l = k~6,Ocutoff~1792, probably owing to the sample size. However, the comparison of the results for l = 7,l = k = 0, l = l = k = 7, Ocutoff = O77000 = 64, and l = l = k = 7, Ocutoff = O00777 = 960 indicates that including small numbers of lower orders of cross terms between polar and Euler angles does not lead to an improvement in performance and sufficient numbers of cross terms are required to improve the performance. This may be one of reasons why Onizuka et al.33 observed worse rather than better performances by taking account of Euler angle dependencies in orientational distributions.
Dependencies of the performance on the cutoff parameters are also examined. In cases of low resolution in which only polar dependencies are taken into account, the effects of the cutoff parameter ccutoff on the recognition performance are not clear for the cases of ccutoff = 0,0.025,0.5. However, in the cases of high resolution the value 0.05 for ccutoff is not small enough to reproduce the orientational distributions for fold recognition. See tables in the auxiliary material52 for details. The threshold ccutoff for significant expansion terms should be set as small as ccutoff~0.025. This is consistent with the fact that as shown in Fig. 2 the mean orientational entropies can be reproduced by employing ccutoff~0.025. Using a value for ccutoff lower than 0.025 does not always yield good performance and may even decrease the recognition power, probably because the expansion terms with small values of coefficients tend to correspond to statistical noise. Thus, the value of 0.025 is used here for ccutoff.
The effects of for a small sample correction are shown in Table III(c). The potential shows a better performance around = 0.2; Naa/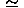 18 000(= 1 467 302/400/0.2). This means that the first digit will be significant in the estimated values of the expansion coefficients for the terms of Olpmplemeke = 1792, because
18 000(= 1 467 302/400/0.2). This means that the first digit will be significant in the estimated values of the expansion coefficients for the terms of Olpmplemeke = 1792, because ![<sub><i>l</i><sub><i>p</i></sub><i>m</i><sub><i>p</i></sub><i>l</i><sub><i>e</i></sub><i>m</i><sub><i>e</i></sub><i>k</i><sub><i>e</i></sub></sub><sup><i>a</i><i>a</i><sup>[prime]</sup></sup>](024901_1-div3_files/024901_1m37.gif) in Eq. (31) becomes about 0.1 for Olpmplemeke = 1792. Thus, the values of = 0.2 and Ocutoff = 1792 would be consistent with one another.
in Eq. (31) becomes about 0.1 for Olpmplemeke = 1792. Thus, the values of = 0.2 and Ocutoff = 1792 would be consistent with one another.
The parameters of l = l = k = 6 with Ocutoff = 1792, ccutoff = 0.025, and = 0.2 are employed here, although Ocutoff = 960 is also good, and could be chosen if one wants to reduce the number of expansion terms. The discrimination of the native structures is successful for 37 of the 79 monomeric decoy sets and for 59 of the 81 immunoglobulin decoy sets using the orientational energy.
The value of ln Pe for each decoy set is shown in Fig. 7; (a) for the decoy sets of monomeric proteins, and (b) for the immunoglobulin decoy sets. The abscissa shows the identification number of the decoy set that is listed for each decoy in tables in the auxiliary material.52 Cross marks and solid lines indicate the values for the case of l = 7,l = k = 0; both are the best case for each decoy set group if only polar-angle dependencies are taken into account. Open circles and broken lines are for the case of l = l = k = 6. For most decoy sets, the performance in the discrimination of the native structures is improved.
 Figure 7.
Figure 7. G. How important are relative orientations between residues in fold recognition?
A summary of the effects for each potential component in Eq. (1) on the performance in fold recognition is listed in Table IV. The energy terms included in the total energy potential are listed in the first column of the table. The performances of those total energy potentials are evaluated by the number of top ranks (no. of tops), the means over all decoy sets of the logarithms of rank probabilities ln Pe in the energy scale and ln Pr in the RMSD scale, and of the Z scores Ze in the energy scale and Zrmsd in the RMSD scale, and the medians of those Z scores in all decoy sets. Also the mean values 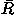 over all decoy sets of the correlation coefficients of rank order between the energies and RMSDs of the decoys are listed for reference.
over all decoy sets of the correlation coefficients of rank order between the energies and RMSDs of the decoys are listed for reference.
First, the results for the monomeric protein decoy set group clearly show the orientational potential eo can achieve a performance comparable to the simple contact potentials, without and with the collapse energy, 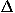 ec and e
ec and e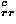 + ec, indicating that residues in the non-native structures are not well positioned with respect to the relative orientation between them.
+ ec, indicating that residues in the non-native structures are not well positioned with respect to the relative orientation between them.
It should be noted here that for the monomeric decoy set group the performance of the contact potential ec without the orientational energy is slightly better than that of the orientational energy eo only, but it is significantly worse for the immunoglobulin decoy set group. Including the collapse energy e causes the performance to become even worse, indicating that the contact potential without the orientational potential does not work at all for these decoy sets. In the case of multimeric proteins, the evaluation of contact energies for residues on the surface of the domain requires other domains and chains to be present. When other domains and chains are not available for a given domain, residue-residue contacts between domains and chains cannot be evaluated. Thus, as already mentioned, unlike short-range potentials, the true ground state of those multimeric proteins in the contact potential requires all of the chains to be present. Especially in the case of immunoglobulin molecules, the interface among constant and variable domains occupies a large portion of the surface of the domains. Thus, the potential consisting of the simple contact energy shows an extremely poor performance for the immunoglobulin decoy sets. On the other hand, the orientational potential only measures how good or bad the relative orientations between contacting residues are, and thus its evaluation does not necessarily require the presence of all domains and chains in multimeric proteins, although it would be more precisely measured if all contacting residues were known; as seen from Eq. (11), the expected value of the orientational energy for contacting residues in native protein structures is adjusted to be equal to zero.
It is noteworthy that in Table IV(a) a large improvement in performance is not seen for the monomeric protein decoy set group, in which decoys have relatively compact structures, by adding the residue-type independent contact energy e to the residue-type dependent contact potential ec except for the case of the energy ec + eo. This fact indicates that optimizing potentials is not simple.
It is interesting to note that the inclusion of the repulsive potential er partially improves the performance for the immunoglobulin decoy set group, in comparison with the case for the monomeric decoy set group. The repulsive potential favors packing densities similar to the residue densities observed in native structures. Thus, the fact that the repulsive potential works well for these decoy sets may indicate that these decoys do not mimic well the native structures with respect to residue density. However, for well designed decoys, the packing potential may work less favorably for the native fold as shown in the case of the monomeric decoy set family.
The performance of the potential function is further improved for both of the present decoy sets by including the simple short-range (,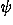 ) potential, strongly indicating that the short-range interactions should not be ignored in fold recognition.
) potential, strongly indicating that the short-range interactions should not be ignored in fold recognition.
The improvement of the performance for fold recognition due to the orientational potential is also observed for almost all decoy sets. In Fig. 8, the value of the logarithms of rank probabilities in the energy scale ln Pe for each decoy set is plotted against the identification number of the decoy set that is listed for each decoy in Table V and tables in the auxiliary material;52 (a) is for the monomeric protein decoy set group and (b) for the immunoglobulin decoy set group. Open circles and broken lines show the values for the potential function that includes the orientational energy eo, and cross marks and solid lines are for the potential without the orientational energy. Even in the decoy sets of the monomeric proteins, ln Pe for each decoy set tends to be more negative in the potential that includes the orientational energy.
 Figure 8.
Figure 8. H. Comparison of the performance of the present potential function with other potentials
The performance of the present potential function for each decoy family is listed in Table V, and that for each decoy set is provided as tables in the auxiliary material.52
Table V and the tables in the auxiliary material52 also show the performances of some of the scoring functions24,25,33,34,35 that have already been tested for some of these decoys. Those scoring functions referred to here are four statistical potentials and one atomic semiempirical potential. These four statistical potentials are the atomic contact potential developed by Samudrala and Moult,13 the distance-dependent pair potential optimized for fold recognition by Toby and Elber,24 the optimal Chebyshev-expanded function -minimizing Z scores devised by Fain, Xia, and Levitt,25 and the distant-dependent angular potential named "3C326" developed by Onizuka et al.33 The atomic semiempirical potential referred to here is a potential based on the CHARMM gas phase implicit hydrogen force field in conjunction with a generalized Born implicit solvation term by Dominy and Brooks,18 which includes specifically a generalized Born, Coulomb, nonpolar solvation and van der Waals energy terms. Data for the potential of Samudrala and Moult13 are taken from Fain, Xia, and Levittet.25
The decoy sets of protein 1FC2 are found in the three decoy set families of fisa, lmds, and lmds_v2, and in all of these decoy sets the present potential failed to identify the native folds. The coordinates of the native fold 1FC2 is for the fragment B of protein A in a complex with immunoglobulin Fc. All chains that interact with the fragment B may be required to estimate the ground state energy for this structure, especially because this fragment is only 43 residues long. The decoy sets of protein 1BBA are also found in two decoy set families, lmds and lmds_v2. This protein is pancreatic hormone that consists of only 36 residues, and is expected to interact with relatively large receptor proteins. Protein 1NKL in lattice_ssfit and semfold can bind lipid, and protein 1BGA8-A in fisa_casp3 is found in the trimeric state in the PDB coordinate file. Thus, one reason why the present potential fails for some decoy sets may be that some chains are missing for the proper estimation of the ground state for these decoy sets. Otherwise, there could be interactions that are not taken into account in the present potential function.
However, overall the present potential function performs well in comparison with other scoring functions. The discrimination for the native structure is successful for 61 of 79 monomeric decoy sets and for 68 of 81 immunoglobulin decoy sets. Also, the mean Z score Ze in the energy scale which is equal to –4.45 for monomeric decoy sets and –3.29 for immunoglobulin decoy sets is statistically significant. For the decoy sets in the globin family hg_structal, interactions between a heme and surrounding residues are not taken into account. Although the present potential fails to identify the native fold for 7 of 29 decoy sets in this family, the RMSD of the lowest energy fold is below 1 Ĺ in 4 of these 7 decoy sets.
Table V clearly shows that the present method outperforms the other potentials for all the decoy families except for the fisa and fisa_casp3 decoy families for which the potential developed by Toby and Elber is better in the mean value of energy Z score, although the present potential performs better than their potential in the cases of 4state_reduced, lattice_ssfit, and lmds decoy families. One of interesting facts is that the atomic semiempirical potential based on the CHARMM potential with a generalized Born, Coulomb, nonpolar solvation and van der Waals energy terms cannot perform better than the present coarse-grained potential, at least for the reported two decoy families 4state_reduced and hg_structal. At the current development stage of atomic potentials, identifying native structures appears to be a hard task, and atomic potentials without explicitly taking account of solvent molecules cannot necessarily perform better than coarse-grained and residue-level statistical potentials. On the other hand, explicitly taking account of water molecules would take too much CPU time to estimate conformational free energies. This fact motivates our studies to develop coarse-grained potentials.
The correlation coefficient R of rank order between the energies and RMSDs of decoys is listed in Table V and tables in the auxiliary material.52 because it was used also in Ref. 25. There are many decoy sets for which the potential succeeds in identifying the native fold and for which both values of Z scores, Ze and Zr, are large but the correlation coefficient R of rank order has values smaller than 0.3; see those values for the decoy set families of lattice_ssfit, lmds, lmds_v2, and semfold. Thus, generally speaking, this measure R may be inappropriate for the evaluation of the performance of scoring functions. It may be appropriate only for some decoy sets, which consist of near-native decoys only, such as the decoy sets in 4state_reduced.
Previous section: II. METHODS
Next section: IV. DISCUSSION
Title Page