The Journal of Chemical Physics, 08 January 2005
J. Chem. Phys. 122, 024901 (2005) (18 pages)
©2005 American Institute of Physics. All rights reserved.
Previous section: III. RESULTS
Next section: REFERENCES
Title Page
IV. DISCUSSION
The present analyses of relative residue-residue orientations clearly indicate that the distribution of residue-residue orientations strongly depends on the Euler angles that specify three degrees of rotational freedom for one residue relative to another, and it is possible to improve the performance of an energy potential in fold recognition by taking account of the Euler angle dependencies in residue-residue orientations.
In the analyses of relative residue-residue orientations by Buchete et al.,34,35 the Euler angle dependencies of residue-residue orientations were not completely taken into account, probably because the number of residue-residue pairs observed in known protein structures is relatively small to reliably estimate the orientational distribution with the required resolution by dividing space into many cells and counting samples observed in each cell. In order to overcome such problems, we chose a method proposed by Onizuka et al.33 in which the observed distribution of residue-residue orientations is represented as a sum of 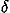 functions each of which represents the observed location in angular space. Then, the distribution of residue-residue orientations is estimated in the expansion with spherical harmonics functions and the coefficients of the expansion terms are estimated by inversely transforming the observed distribution represented as the sum of functions.
functions each of which represents the observed location in angular space. Then, the distribution of residue-residue orientations is estimated in the expansion with spherical harmonics functions and the coefficients of the expansion terms are estimated by inversely transforming the observed distribution represented as the sum of functions.
High frequency modes in the expansion must be ignored because they reflect artificial contributions originating in the small size of samples. Each term in the expansion has a different resolution with various combinations of frequencies for each coordinate axis. A trivial example is that the first term g00000 corresponding to a uniform distribution has the lowest resolution. Here, resolution of each term is represented by Olpmplemeke, that is, defined as the number of frequency modes lower than or equal to (lp, mp, le, me, ke) by Eq. 32 and only terms whose Olpmplemeke is less than a cutoff value Ocutoff are used. The merit of this method is that the distribution can be constructed by using only expansion terms whose resolutions are low enough to be able to be estimated from a limited number of samples of known protein structures. On the other hand, the cell partitioning method has a fixed resolution for each coordinate axis, so that high frequency modes with large values of Olpmplemeke can be included in the estimation of orientational distributions.
Because the resolution of each term is different from others, each term is differently corrected for the small size of samples according to its resolution; see Eqs. (27,28,29,30,31,32) In this correction scheme, the number of residue-residue pairs required for the estimation of an expansion coefficient clpmplemeke increases proportionally with its resolution Olpmplemeke. The proportionality constant was determined on the basis of the performance of the potentials in fold recognition. Also, the maximum resolution that can be estimated depends on the sample size. The maximum values for lp, mp, le, me, and ke, and for Olpmplemeke are determined on the basis of the performance of the potentials in fold recognition.
Also, the reference distribution of residue-residue orientations for the present orientational potentials is not the overall distribution for all types of amino acid pairs but the uniform distribution, differing from other works.33,34,35 It depends on decoy sets whether the uniform distribution for a reference distribution is effective. If the structures of decoys have a similar overall distribution to that of native structures, then it will not be effective. However, such an overall distribution of residue-residue orientations would not be intrinsically characteristic of non-native conformations but instead of native structures of proteins. If so, this overall distribution may be one of the important characteristics to distinguish protein-like structures from others. On the other hand, there is no reason to avoid employing the uniform distribution for a reference distribution. The use of the uniform distribution as a reference distribution is desirable to fully evaluate the orientational distribution of each type of contacting residue pair in decoy structures. Our scheme differs from previous works33,34,35 and allows us to more properly evaluate the effectiveness of the orientational potential on fold recognition.
However, the present method of evaluating orientational energies between contacting residues requires the evaluation of a large number of expansion terms.53 Although this feature is a trade-off accompanied with the simplification of representing residues by single points, it can be an obstacle to using this method in CPU intensive calculations in which energy evaluations of many conformations are required. To reduce CPU time in the evaluation of orientational energies, orientational energies could be precalculated at grid points in the polar and Euler angular space, although this approach requires a large memory and disk space as a trade-off against CPU time.
In the present work, the total energy in Eq. (1) is assumed to consist of a simple sum of energy terms, because each energy potential has been evaluated in a similar manner as the potential of mean force from statistical distributions of residues observed in protein structures, avoiding overcounting particular interactions. One might assume a different weight for each contribution to the total energy, and try to optimize a weight for each energy term by minimizing the Z score Ze for the decoy sets.16 However, equal weights are employed here for each term, because a set of optimum weights could strongly depend on the training decoy sets. For example, if bad contacts are removed and torsion angles are optimized for decoy structures, then the packing potential and the secondary structure potential tend to be useless in discriminating the native structures from decoys, and optimum weights for those potentials determined by minimizing the Z score would take on relatively small values. The training decoys for optimizing a weight of each energy term in a total potential must be carefully generated without bias. In addition, generating unfolded decoys is also necessary to obtain an appropriate value with such an optimizion method for the collapse energy, which is represented as e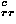 and which is an extremely important energy for a protein to fold that compensates for the large conformational entropy loss of compact conformations.
and which is an extremely important energy for a protein to fold that compensates for the large conformational entropy loss of compact conformations.
Previous section: III. RESULTS
Next section: REFERENCES
Title Page